Malformaciones Renales
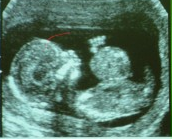
¿Qué es un ONFALOCELE?
El Onfalocele o Exonfalo es un defecto en el desarrollo de la pared abdominal, en el que las vísceras herniadas están cubiertas por peritoneo, amnios y gelatina de Wharton. Los órganos comprometidos en la evisceración pueden ser asas de intestino delgado o grueso, hígado y estómago, estando siempre incluido el cordón en el saco herniado. Se diagnostica a partir de la semana 12 de gestación, ya que durante la vida fetal el intestino medio migra dentro del cordón umbilical, para retornar a la cavidad alrededor de las semanas 10-12. El defecto es consecuencia de un fallo en el retorno de este intestino, quedándose fuera de la cavidad abdominal, recubierto por peritoneo y membrana amniótica, quedando el cordón umbilical inserto en este saco que cubre el defecto. Es una malformación congénita relativamente común, estimándose su incidencia en 1/4.000 nacimientos. Casi la mitad de los pacientes con onfalocele presenta otras malformaciones asociadas, principalmente malformaciones cardíacas, gastrointestinales, genitourinarias, musculoesqueléticas y del sistema nervioso central, además de asociarse a Trisomñias 13,18 y 21, además de formar parte de síndromes polimalformativos como el Síndrome de Beckwith-Wiedemann, que fue este caso.
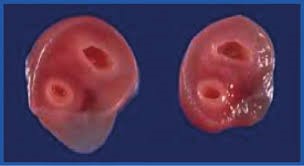
ARTERIA UMBILICAL ÚNICA (AUU)
La arteria umbilical única se caracteriza por tener el cordón umbilical solo una arteria en lugar de dos. Es una malformación congénita con una incidencia estimada de entre el 0,5–0,8% de los nacimientos únicos y se asocia con anomalías congénitas en entre el 19–66% de los casos. Los sistemas más afectados son el genitourinario, el cardiovascular, el digestivo y el osteoarticular en la siguiente proporción: aparato genitourinario (18,4%), las malformaciones de extremidades (15,4%), las gastrointestinales (12,3%), las cardiovasculares (10,7%), y las del sistema nervioso central (7,7%), acompaña con frecuencia de crecimiento intrauterino restringido (CIR). Se asocia con trisomías 13, 18 y 21, y con otras anomalías cromosómicas.
En la embriología del desarrollo el cordón nefrógeno, del cual deriva el cordón umbilical, da lugar al aparato urinario y gonadal.
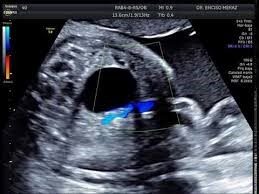
AGENESIA RENAL.
Es una malformación congénita poco frecuente del tracto renal que se caracteriza por la ausencia total de desarrollo de uno o ambos riñones (agenesia renal uni- o bilateral, respectivamente), acompañada de ausencia de uréter(es). La prevalencia de la agenesia renal unilateral (AR) al nacimiento se estima en 1/2.000 aproximadamente, mientras que la prevalencia fetal de la agenesia renal bilateral en Europa se ha estimado en 1/8.500.
La diabetes mellitus materna o el uso de fármacos específicos durante el embarazo también pueden ocasionar agenesia renal. La mayoría son unilaterales, afectando más frecuentemente a varones y riñón izquierdo con hipertrofia compensadora en el riñón contralateral sano. Se asocia con anomalías genitales, renales o ureterales contralaterales, siendo el reflujo vésico-ureteral (RVU) lo más frecuente (25% de los casos)(2).
El diagnóstico se basa en la ecografía, que muestra una fosa renal vacía y ausencia de riñón ectópico. Los exámenes radiológicos adicionales como la resonancia magnética (RM) permiten confirmar el diagnóstico. El riñón contralateral puede experimentar hipertrofia compensadora.
En los casos familiares, la AR unilateral se hereda típicamente de modoautosómico dominante con penetrancia incompleta. La AR bilateral se transmite con herencia autosómica recesiva.
SÍNDROME DE BECKWITH-WIEDEMANN
El SBW está causado por alteraciones en genes del cromosoma 11p15.5. Se ha descrito un pequeño porcentaje de casos de trasmisión familiar. Puede ser recurrente en una misma pareja y presentar diferentes espectros clínicos. Su incidencia se estima en 1 cada 13.700 nacidos vivos, siendo de distribución igual por sexos.
Puede acarrear hemihiperplasia y/o macroglosia (ocasionando dificultades en alimentación y habla). Aparece hipoglucemia en el 30-50% de los recién nacidos. Es común una forma facial reconocible, a menudo normalizada en la edad adulta. Además de macrosomia, macroglosia, hemihiperplasia e hipoglucemia, los hallazgos pueden incluir onfalocele, hernia umbilical, diástasis de rectos, tumor embrionario, pliegues del lóbulo de la oreja y hoyos helicoidales, nevos flamígeros, malformaciones vasculares, visceromegalia implicando órganos abdominales, citomegalia adrenocortical fetal (patognomónico), anomalías renales, historia familiar positiva y, en raras ocasiones, paladar hendido. Existen malformaciones cardíacas en el 9-34% de los casos y la mitad de éstos presentan una cardiomegalia que desaparece de forma espontánea. Las miocardiopatías son raras. Los pacientes tienen mucha predisposición a tumores malignos embrionarios principalmente en los primeros 8 años de vida, con un riesgo estimado de 7,5% (en un rango del 4-21%).